A genom nyelvének feltörése
A gépi tanulási modelleket szerteágazó, óriási mennyiségű adattal táplálják: két emblematikus forrás az ENCODE (A DNS-elemek enciklopédiája – Encyclopedia of DNA Elements) és a GTEx (Genotípus–szövet expresszió – Genotype-Tissue Expression) projekt, amelyek maguk is áttörést hoztak a humán genom megértésében. Az ENCODE célja eredetileg az volt, hogy feltérképezze, pontosan mely szakaszai “működnek” genomunknak: 20%-ukról már tudjuk, hogy szabályozó vagy egyéb biológiai funkcióval rendelkeznek, szemben a korábban “szemét-DNS”-nek hitt nagy többséggel. Lényeges, hogy ezek a régiók döntő szerepet játszhatnak abban, hogyan alakulnak ki betegségek, hiszen a genetikai eltérések többsége éppen itt található.
A GTEx erre az alapra épített – egyre több olyan génvariánst találtak, amelynek nem ismerték a szerepét, hiszen nem a kódoló génekben fordultak elő, hanem az ismeretlen tartományokban. Ezért kezdte el a projekt feltérképezni, hogy egyes variánsok hogyan befolyásolják a génkifejeződést különböző szövetekben vagy sejttípusokban, vagyis működésbe lépnek-e, aktívak maradnak-e vagy éppen lezáródnak.
Az adatkincsek továbbfejlesztik az MI-t
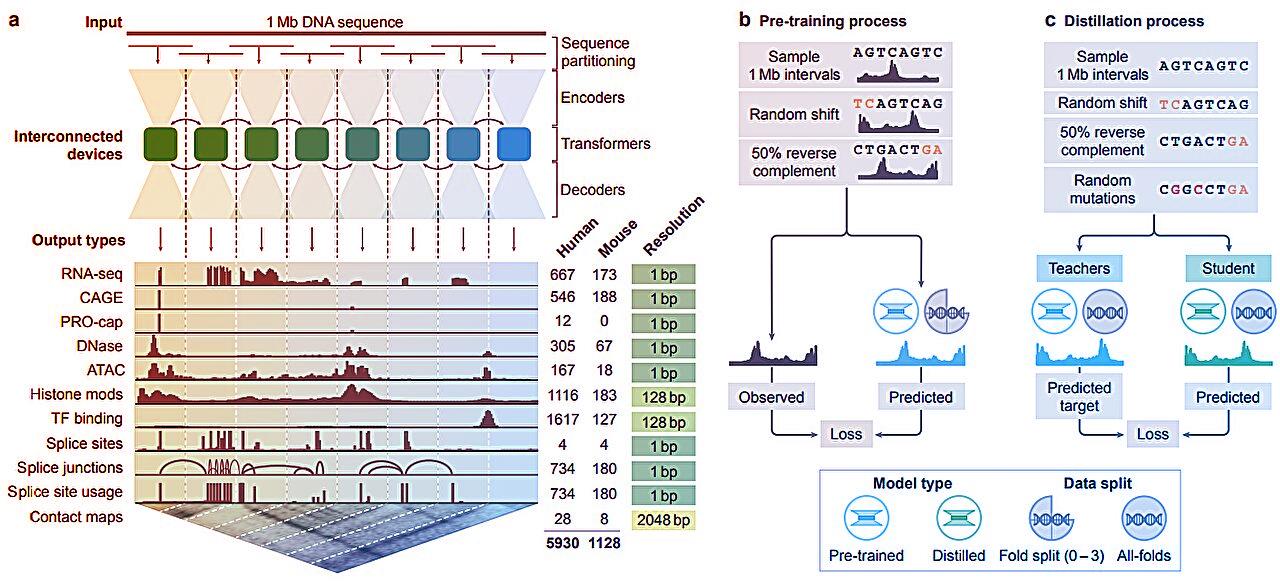
Az ENCODE és a GTEx szabadon hozzáférhető, közösségi adatbázisokként születtek, amelyek immár évtizedekkel az indulásuk után is új MI-modellek kibontakozásához vezetnek. Ezek a hatalmas, megbízható adathalmazok tették lehetővé, hogy az AlphaGenome-hoz hasonló modern modellek egyre pontosabb előrejelzéseket adjanak arra vonatkozóan, miként szabályozódnak a gének. Mindazonáltal minél mélyebb, sejtszintű és zavaró (perturbációs) adatokat szerzünk, annál pontosabbá válhatnak ezek az előrejelzések.
Számos új projekt, például az Emberi Sejtatlasz (Human Cell Atlas) vagy a Génszabályozási Megfigyelő (Gene Regulation Observatory), éppen abban segít, hogy részletesen feltérképezzük, hogyan működnek a sejtek és szövetek egészséges és beteg állapotban.
Az MI szerepe a genom szabályozásának feltárásában
Ma már vezető laboratóriumok dolgoznak azon, hogy MI segítségével megfejtsék a génszabályozás “forráskódját”. Ilyen például a Broad Intézet Epigenomika Programja, ahol kizárólag gépi tanulással elemzik, hogy a génekhez közel eső szabályozó elemek (mint a promoterek) mikor és milyen sejtdifferenciáció során aktiválódnak. A kutatók ma már a 3D-s genomstruktúra elemzésére is MI-t alkalmaznak, hogy jobban megértsék a génkifejeződést távolról irányító kapcsolatrendszereket. Mindezt azért, hogy minél többféle sejttípusban, akár mutációkkal mesterségesen megzavart genomokon is tudják tesztelni és tanítani a modelleket.
A következő lépések: még több és még részletesebb adat
Ahhoz, hogy az MI teljes kapacitását kiaknázzuk a genomika területén, új típusú, célzott adatokat kell létrehoznunk. Például nemcsak arra van szükség, hogy tudjuk, hol milyen génszabályozó fehérjék kötődnek a DNS-hez, hanem arra is, hogy a különböző variánsok és zavaró hatások (pl. betegségek) milyen folyamatokat indítanak el egy-egy sejttípusban vagy szövetben.
Fontos, hogy ezeknek az adatforrásoknak együttesen kell kiszolgálniuk az MI-modellek igényeit, hogy ne csak önálló variánsokat elemezzünk, hanem átfogó, rendszerszintű szabályokat alkothassunk a génkifejeződésről és az öröklődő betegségek hátteréről is.
Az MI a pontosabb genetikai diagnózisok kulcsa lehet
Az elérhető, egyre pontosabb adatok és a csúcskategóriás MI-modellek lehetőséget teremtenek arra, hogy a genetikai szűrések során talált, eddig megmagyarázhatatlan variánsokat is jobban megértsük. Ez segíthet a súlyos betegségek gyorsabb felismerésében és személyre szabottabb kezelésében. Az AlphaGenome-hoz hasonló rendszerek révén végre közelebb kerülhetünk egy évtizedes vitához: vajon egyedi variánsokat érdemes vizsgálni, vagy inkább átfogó MI-modellekre bízzuk a szabályok feltárását?
Mindezt figyelembe véve a genomika jövője a minél részletesebb, nyíltan hozzáférhető adatbázisokon, valamint a fejlődő mesterséges intelligencián múlik. Ha jól használjuk ezeket, teljesen új dimenziókat tárhatnak fel előttünk az emberi egészség és betegségek megértésében.